
再生医療や遺伝子治療の技術革新が進む中、開発中のシーズが「再生医療等製品」に該当するかどうかの判断は、その後の開発ロードマップや薬事戦略を決定づける極めて重要なステップです。薬機法における定義は一見複雑に見えますが、その本質を理解することで、適切な規制要件への対応や、条件付き期限付承認制度といったメリットの活用が可能になります。
この記事では、製薬企業やバイオベンチャーの研究開発・薬事担当の方々に向けて、薬機法第2条第9項に基づく再生医療等製品の定義をわかりやすく解説します。医薬品や医療機器との境界線、該当性判断のポイント、そして開発におけるメリットまで、実務に役立つ情報を網羅的に整理しました。自社製品のポジショニングを明確にし、最短ルートでの実用化を目指すための一助としてお役立てください。
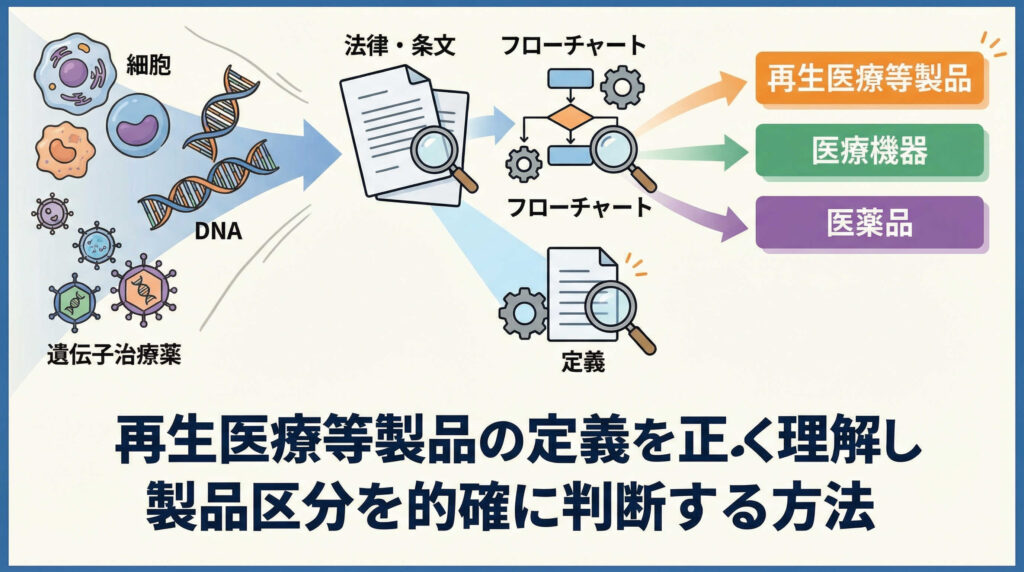
再生医療等製品の定義とは【薬機法第2条第9項の解説】

再生医療等製品という区分は、平成25年の薬事法改正(現・薬機法)によって新たに設けられたカテゴリーです。従来の医薬品や医療機器の枠組みでは評価が難しかった、生きた細胞や遺伝子を用いる製品の特性に合わせて定義されています。
その法的根拠となるのが薬機法第2条第9項です。ここでは、大きく分けて「細胞加工製品」と「遺伝子治療用製品」の2つが定義されています。条文の構造を理解することが、該当性判断の第一歩となりますので、それぞれの要件を詳しく見ていきましょう。
第1号:人又は動物の細胞に培養等の加工を施したもの
第1号では、人または動物の細胞に「培養」などの加工を施したものが規定されています。ポイントは、単に細胞を取り出しただけでなく、人工的な操作(加工)が加えられている点です。
具体的には、以下のような目的を持つものが該当します。
- 身体の構造・機能の再建、修復または形成
- 疾病の治療または予防
例えば、iPS細胞やES細胞由来の分化細胞、体性幹細胞製品、あるいは免疫細胞療法に用いる加工細胞などがこれに含まれます。「生きた細胞」が主役となって治療効果を発揮するものが、この第1号のカテゴリーとなります。
第2号:人又は動物の細胞に導入され、体内で発現する遺伝子治療用製品
第2号は、いわゆる遺伝子治療用製品を指します。これは、人または動物の細胞に導入され、体内で発現する遺伝子を含有する医薬品等を意味します。
ここでの特徴は、細胞そのものではなく、「遺伝子発現構成体(ベクターなど)」が製品の主体であるケースが多い点です。
- ウイルスベクター(AAV、レンチウイルス等)を用いたIn vivo遺伝子治療薬
- プラスミドDNAを用いた製品
これらは、体内で特定のタンパク質を作らせたり、欠損している遺伝子を補ったりすることで治療効果を狙います。細胞加工製品とは異なり、遺伝子の導入と発現が定義の核となっています。
法的定義における「加工」の範囲と判断基準
定義の中で特に議論になりやすいのが、「どこからが加工にあたるのか」という境界線です。薬機法上、単なる分離や洗浄、保存といった細胞の性質を変えない操作は「加工」とはみなされず、輸血用血液製剤などの範疇になることがあります。
一方で、「細胞の生物学的特性、生理学的機能を変化させる操作」は加工とみなされます。
- 加工に該当する例: 培養による増殖、薬剤処理による活性化、分化誘導、遺伝子導入など
- 加工に該当しない例: 組織の細断、遠心分離、洗浄、凍結保存、抗生物質処理など
この「加工」の有無が、再生医療等製品かどうかの大きな分かれ道となるのです。
再生医療等製品の該当性判断における重要ポイント

条文上の定義を確認しただけでは、実際の製品がどのカテゴリーに当てはまるか判断に迷うケースも少なくありません。特に新規性の高いモダリティの場合、その作用機序や使用目的が判断の鍵を握ります。
再生医療等製品の該当性を判断する際は、単に「細胞を使っているか」「遺伝子を使っているか」という物質的な側面だけでなく、「何を目的として使用されるか」という意図も重要視されます。ここでは、判断において特に重要となる3つの視点を深掘りします。
身体の構造・機能の再建・修復・形成を目的とする場合
一つ目の重要な判断基準は、製品の使用目的が「身体の構造や機能の再建・修復・形成」にあるかどうかです。これは、従来の低分子医薬品が主に症状の緩和や病原体の排除を目的としていたのに対し、再生医療等製品は「失われた組織そのものを補う」という根本的な治療を目指す点に特徴があります。
例えば、軟骨欠損に対する培養軟骨移植や、重症熱傷に対する培養皮膚などが典型例です。このように、細胞や組織そのものが生着し、身体の一部として機能することが期待される場合は、再生医療等製品としての該当性が高まります。
疾病の治療・予防を目的とする場合
二つ目は、「疾病の治療または予防」を目的とする場合です。これは主に、がん免疫療法や遺伝子治療において重要な観点となります。組織の修復そのものではなく、細胞や遺伝子の働きによって病気を治すケースです。
例えば、CAR-T細胞療法のように、遺伝子改変したT細胞を投与してがん細胞を攻撃させる場合、構造の修復というよりは「治療」が主目的となります。しかし、高度な加工が施された細胞を用いているため、これも再生医療等製品(第1号および第2号の要素を含む製品)として扱われます。作用機序が薬理学的であっても、主体が加工細胞や遺伝子導入であれば、こちらの区分になるのです。
ヒト細胞加工製品と動物細胞加工製品の分類
三つ目は、原材料となる細胞の由来です。再生医療等製品は、「ヒト細胞加工製品」と「動物細胞加工製品」に大別されます。
- ヒト細胞加工製品: 自己(自家)または同種(他家)のヒト細胞由来。倫理的な配慮や感染症スクリーニングが重要。
- 動物細胞加工製品: 動物の細胞由来。異種移植に伴う拒絶反応や未知のウイルス感染リスク(異種感染症)への厳格な管理が必要。
どちらに該当するかによって、求められる安全性試験の項目や品質管理基準(GCTPの運用)が異なりますので、開発初期段階で明確にしておく必要があります。
再生医療等製品と他カテゴリーとの区分・境界線

開発戦略を立てる上で最も悩ましいのが、医薬品や医療機器との境界領域にある製品(ボーダーライン製品)の扱いです。製品区分が変われば、適用される規制や承認審査のプロセスがガラリと変わってしまいます。
ここでは、間違いやすい他カテゴリーとの違いや、判別のための考え方を整理します。PMDAへの相談前に、自社製品がどのカテゴリーに近いのか、論理的に整理しておきましょう。
医薬品(生物由来製品)との違い
最も混同しやすいのが、生物由来製品(医薬品)との区別です。ワクチンや血液製剤も生物由来ですが、これらは一般的に医薬品として扱われます。
決定的な違いは、「期待される作用の主役が何か」という点にあります。
- 再生医療等製品: 生きた細胞そのもの、あるいは導入された遺伝子が体内で機能・発現することで効果を発揮する。
- 医薬品(生物由来製品): 細胞から抽出・精製されたタンパク質(抗体、酵素、サイトカインなど)や、細胞を含まないエクソソームなどが主成分である。
つまり、最終製品に「生きた細胞」が含まれているか、あるいは「遺伝子治療用ベクター」であるかが主要な判断基準となります。
医療機器およびコンビネーション製品との判別
細胞や組織をスキャフォールド(足場材料)と組み合わせて使用する場合、医療機器なのか再生医療等製品なのか判断が難しくなります。このような製品は「コンビネーション製品」と呼ばれます。
この場合、「主たる機能を発揮するのがどちらか」で判断されます。
- 再生医療等製品: 細胞が生着・増殖・分化することで治療効果の主役を担う場合(例:細胞シート)。
- 医療機器: 細胞は補助的な役割で、あくまで人工材料による構造支持や置換が主目的である場合。
どちらの要素が治療効果の本質を担っているかを科学的に説明できることが重要です。
輸血用血液製剤や移植用臓器との区別
輸血用血液製剤や移植用臓器も、生きた細胞や組織を含みますが、これらは再生医療等製品には含まれません。
- 輸血用血液製剤: 加工の程度が低く、歴史的に医薬品として管理されてきた経緯から、医薬品とみなされます。
- 移植用臓器: 臓器移植法に基づき医療行為として行われるものであり、製品として流通させて販売するものではないため、薬機法の対象外です。
「高度な加工が施されているか」「産業として流通させる製品か」という点が、これらのカテゴリーとの分水嶺となります。
薬機法と再生医療等の安全性の確保等に関する法律(安確法)の適用範囲の違い
最後に、よくある誤解として「再生医療等の安全性の確保等に関する法律(安確法)」との混同があります。
- 薬機法(再生医療等製品): 企業が製造販売承認を取得し、製品として広く市場に流通させるもの。保険収載を目指すルート。
- 安確法(特定細胞加工物等): 医療機関が主体となり、自由診療や臨床研究として特定の患者さんに施術するもの。
開発中のシーズを「製品として売り出す」のか、「医療技術として提供する」のかによって、適用される法律が全く異なります。本記事で解説しているのは、前者の薬機法に基づく製品の定義です。
再生医療等製品に分類されることによる開発・薬事戦略上のメリット

再生医療等製品の定義に該当すると判断されることは、単に規制対応が厳格になるという側面だけではありません。実は、開発企業様にとっては、他のカテゴリーにはない柔軟な制度を活用できる好機でもあります。
国は再生医療の早期実用化を強力に推進しており、世界に先駆けて「条件付き・期限付承認制度」を導入しました。ここでは、再生医療等製品だからこそ享受できる開発・薬事戦略上のメリットとして、早期の市場投入を可能にする「条件付き・期限付承認制度」、優先的な審査を受けられる「先駆け審査指定制度」、そして市場独占性に関わる「再審査期間」の3つの大きな利点について具体的に解説します。
条件付き期限付承認制度の活用
再生医療等製品の最大の特徴とも言えるのが、「条件付き期限付承認制度」です。これは、臨床試験で「有効性が推定」され「安全性が確認」できれば、特別に早期承認を与えるという制度です。
通常の医薬品では検証的臨床試験(第Ⅲ相試験)で確実な有効性の証明が求められますが、再生医療等製品は症例数を集めるのが難しいケースが多いため、市販後にデータを収集・評価することを条件に、早期に市場へ投入することが可能です。これにより、開発期間の大幅な短縮と早期の収益化が期待できます。
先駆け審査指定制度の対象可能性
画期的な治療薬を世界に先駆けて実用化するための「先駆け審査指定制度」の対象となる可能性があります。この指定を受けると、以下のような優遇措置が受けられます。
- 優先相談: PMDAとの相談待ち時間が短縮される。
- 事前評価の充実: 承認申請前からデータを随時提出し、審査を進められる。
- 優先審査: 審査期間が通常よりも大幅に短縮される(目標審査期間が6ヶ月など)。
特に再生医療分野は国際競争が激しいため、この制度の活用は大きなアドバンテージとなります。
希少疾病用再生医療等製品の指定制度
対象患者数が少ない(国内で5万人未満など)難病治療用の製品であれば、「希少疾病用再生医療等製品」の指定を受けられる可能性があります。いわゆるオーファン指定です。
指定を受けると、助成金の交付、税制措置、指導・助言の優先的な実施に加え、再審査期間の延長などのメリットがあります。ニッチな疾患をターゲットにすることの多い再生医療ベンチャーにとって、開発コストのリスクを低減する重要な制度と言えるでしょう。
製品開発における規制要件と品質管理基準

再生医療等製品として開発を進める場合、従来の医薬品(GMP)とは異なる、特有の品質管理基準や規制要件に対応する必要があります。細胞という「生き物」を扱う難しさが、ここに集約されています。
開発初期からこれらの規制要件を理解し、データパッケージを構築していくことが、後の承認審査をスムーズに進める鍵となります。ここでは、特に留意すべき3つのポイントを解説します。
GCTP省令(Good Gene, Cellular, and Tissue-based Products Manufacturing Practice)への適合
再生医療等製品の製造管理および品質管理には、GCTP(Good Gene, Cellular, and Tissue-based Products Manufacturing Practice)省令が適用されます。
従来のGMPとの大きな違いは、原材料(細胞)の不均一性を前提とした管理が求められる点です。
- ドナーごとの個体差への対応
- 無菌操作の徹底(最終滅菌ができないため)
- バリデーション(検証)とベリフィケーション(確認)の使い分け
これらを適切に文書化し、運用する体制を構築することが、製造業許可および承認取得の必須条件となります。
非臨床試験における安全性評価の考え方
非臨床試験(動物実験等)においても、独特の考え方が必要です。ヒトの細胞を動物に投与しても、免疫拒絶で正しく評価できない場合があるためです。
特に重要視されるのが「造腫瘍性(がん化リスク)」の評価です。加工した細胞が体内で腫瘍を作らないか、目的外の部位に分布しないか(体内分布)を慎重に確認する必要があります。従来の毒性試験のパッケージをそのまま適用するのではなく、製品の特性に応じた「科学的に合理的な試験デザイン」を組むことが求められます。
治験(臨床試験)デザインの特性と留意点
治験(臨床試験)のデザインも、低分子医薬品とは異なります。二重盲検試験(プラセボ対照)が倫理的・技術的に難しいケースが多いためです(例:開頭手術が必要な脳内投与など)。
そのため、過去の診療録(ヒストリカルコントロール)を対照群として設定したり、少数の症例で有効性を評価したりする工夫が必要です。統計的な有意差を出すことが難しい場合でも、臨床的な意義(Clinical Relevance)をどう説明するかが、承認審査の焦点となります。
製品区分・定義の判断に迷った際のアクションプラン

ここまで解説してきた通り、再生医療等製品の定義や区分は非常に専門的で、個別具体的な判断が求められます。「たぶんこれだろう」という自己判断で開発を進めると、後になって手戻りが発生し、莫大な時間とコストを失うリスクがあります。
確実な開発を進めるためには、専門機関との対話が欠かせません。判断に迷った際に取るべき具体的なアクションプランを提示します。
PMDA(医薬品医療機器総合機構)のRS戦略相談の活用
最も確実なのは、規制当局であるPMDA(医薬品医療機器総合機構)の相談制度を活用することです。特に「RS(レギュラトリーサイエンス)戦略相談」は、開発初期段階からシーズの製品区分や必要な試験項目について指導・助言を受けられる枠組みです。
ここで「再生医療等製品に該当する」という見解や、他カテゴリーとの境界に関する合意形成を得ておくことで、その後の開発を安心して進めることができます。公式な議事録は、開発の羅針盤となります。
事前面談による製品該当性の確認プロセス
RS戦略相談を申し込む前段階として、「事前面談」を活用することをお勧めします。これは、正式な相談の前に、相談内容の整理や資料の過不足を確認するための手続きです。
いきなり本番の相談に臨むと、論点がずれていたり、準備不足で有益な回答が得られなかったりすることがあります。事前面談を通じてPMDA担当者と認識をすり合わせ、こちらの質問意図を明確に伝えておくことが、有意義な相談結果を引き出すコツです。
専門のCRO・CDMOへの相談による開発ロードマップ策定
社内に薬事の専門家がいない場合は、再生医療分野に強いCRO(開発業務受託機関)やCDMO(医薬品開発製造受託機関)へ相談するのも有効な手段です。
彼らは多くの企業の開発支援を行っており、類似製品の事例や最新の規制動向に精通しています。PMDA相談に向けた資料作成のサポートや、現実的な開発ロードマップの策定支援を受けることで、社内リソースを研究開発そのものに集中させることができます。外部の知見をうまく取り入れることが、成功への近道と言えるでしょう。
まとめ

再生医療等製品の定義は、薬機法第2条第9項に基づき、「細胞への加工」や「遺伝子発現」を核として規定されています。医薬品や医療機器との境界は、製品の「使用目的」や「作用の主役」によって判断されます。
この区分に該当することで、GCTPなどの厳しい品質管理が求められる反面、条件付き期限付承認制度などの強力な開発メリットを享受できる可能性があります。
重要なのは、自己判断で完結させず、開発の早い段階でPMDAや専門家と連携し、製品の該当性を明確にすることです。正確な定義の理解と適切な区分の特定こそが、革新的な治療を患者さんに届けるための最短ルートとなるでしょう。
再生医療等製品の定義についてよくある質問

再生医療等製品の定義や区分に関して、実務担当者の方からよく寄せられる質問をまとめました。細かい判断に迷った際の参考にしてください。
- Q1. 細胞を洗浄・保存しただけの場合も「加工」にあたり、再生医療等製品になりますか?
- Q2. 患者さんの体外で遺伝子を導入した細胞を戻す場合(Ex vivo)は、第1号と第2号のどちらですか?
- Q3. 自由診療で使われている細胞加工物と、再生医療等製品は何が違うのですか?
- Q4. 海外(欧米)での分類と日本の再生医療等製品の定義は同じですか?
- Q5. 再生医療等製品の承認までの期間はどのくらいですか?
{
"@context": "https://schema.org",
"@type": "FAQPage",
"mainEntity": [
{
"@type": "Question",
"name": "細胞を洗浄・保存しただけの場合も「加工」にあたり、再生医療等製品になりますか?",
"acceptedAnswer": {
"@type": "Answer",
"text": "いいえ、原則としてなりません。薬機法上の「加工」とは、細胞の生物学的特性や生理学的機能を変化させる操作(培養、分化誘導、遺伝子導入など)を指します。単なる分離、組織の細断、洗浄、凍結保存などは加工に含まれず、輸血用血液製剤や移植医療の範疇として扱われることが一般的です。"
}
},
{
"@type": "Question",
"name": "患者さんの体外で遺伝子を導入した細胞を戻す場合(Ex vivo)は、第1号と第2号のどちらですか?",
"acceptedAnswer": {
"@type": "Answer",
"text": "基本的には「第1号(細胞加工製品)」としての性格も持ちますが、遺伝子治療用製品としての側面が強いため、実務上は「遺伝子治療用製品(第2号)」に準じた規制や評価が求められることが多いです。ただし、定義上は「遺伝子導入細胞」として第1号に含まれると解釈されるケースもあり、製品の特性によるためPMDAへの確認が必須です。"
}
},
{
"@type": "Question",
"name": "自由診療で使われている細胞加工物と、再生医療等製品は何が違うのですか?",
"acceptedAnswer": {
"@type": "Answer",
"text": "適用される法律と目的が異なります。自由診療や臨床研究で使われるものは「特定細胞加工物」と呼ばれ、「安確法」の下で医療機関が主体となって提供する医療技術です。一方、「再生医療等製品」は「薬機法」の下で企業が製造販売承認を取得し、製品として広く流通・販売するものを指します。"
}
},
{
"@type": "Question",
"name": "海外(欧米)での分類と日本の再生医療等製品の定義は同じですか?",
"acceptedAnswer": {
"@type": "Answer",
"text": "概念は近いですが、法的な枠組みは異なります。欧州ではATMP(先進医療医薬品)、米国では細胞・遺伝子治療製品(CBER管轄の生物製剤)として分類されます。日本独自の「再生医療等製品」という独立したカテゴリーや「条件付き期限付承認制度」は海外にはない特徴的な仕組みですので、グローバル開発の際は各国の規制とのギャップ分析が必要です。"
}
},
{
"@type": "Question",
"name": "再生医療等製品の承認までの期間はどのくらいですか?",
"acceptedAnswer": {
"@type": "Answer",
"text": "品目によりますが、条件付き期限付承認制度や先駆け審査指定制度を活用することで、通常の医薬品よりも短縮される傾向にあります。優先審査の場合、申請から承認までの目標審査期間は6ヶ月〜9ヶ月程度とされていますが、申請前の相談やデータ準備の期間を含めると、開発開始から数年は要するのが一般的です。"
}
}
]
}